A RARE CASE OF POMPES DISEASE PRESENTED AS FLOPPY INFANT
Abstract
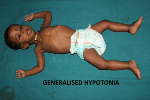
Glycogen storage disease (GSD) are due to mutation of genes that codes for proteins involved in glycogen synthesis.Pompes disease ,also referred as acid maltase deficiency or glycogen storage disease type is an autosomal recessive disorder caused by a deficiencyof lysosomal enzyme acid glucosidase(GAA).1 It was the first recognised lysosomal storage disease and is the only glycogen storage disease that is also a lysosomal storage disease .In pompes disease,lysosomal glycogen accumulates in many tissues , with skeletal ,cardiac and smooth muscles prominently involved2 3.We report a case of pompes disease ,occured in 11 months old male child presented with floppiness of limbs and motor devel opmental delay since 5 months of age.This was one of the very rare cases reported in India.
Full Text:
PDFReferences
Hers HG. Alpha-Glucosidase deficiency in generalized glycogen storage disease (Pompe’s disease). Biochem J 1963;86:11–16.
Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Beaudet A, Scriver C, SlyWet al. eds. The Metabolic and Molecular Bases of Inherited Disease.NewYork: McGraw Hill, 2001:3389–3420.
Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr 2004;144:S35–S43. 4. Pompe JC. Over idiopatische hypertrophie van het hart. Ned Tijdshr Geneeskd1932;76:304.
van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332–340.
Kishnani P, Hwu W, Mandel H, Nicolino M, et al. On behalf of the Infantile-onset Pompe Disease Natural History Study Group a retrospective, multinational, multicentre study on the natural history of infantile-onset Pompe disease. J Peds.
Slonim AE, Bulone L, Ritz S, Goldberg T, et al. Identification of two subtypes of infantile acid maltase deficiency. J Pediatr 2000;137:283–285.
Umapathysivam K, Hopwood JJ, Meikle PJ. Determination of acid-glucosidase activity in blood spots as a diagnostic test for Pompe disease. Clin Chem 2001;47:1378–1383.
Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta 2004;347:97–102.
Niizawa G, Blanco M, Casentini C, Borrajo G, et al. Newborn screening for MPS1 and Pompe disease: a pre-pilot study. Mol Genet Metab 2005;84:A202.
Li Y, Scott CR, Chamoles NA, Ghavami A, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem 2004;50:1785–1796.
Zhang H, Kallwass H, Young S, Carr C, et al. A comparison of methods for lysosomal acid—glucosidase activity determination from dried blood spots for diagnosis of Pompe disease. Genet Med. 2006;8:302–306.
Nelson textbook of paediatrics 19 th edition page 499-500
Refbacks
- There are currently no refbacks.

This work is licensed under a Creative Commons Attribution-NoDerivatives 4.0 International License.
An initiative of The Tamil Nadu Dr M.G.R. Medical University